一、 研究背景:
两相反应是电极材料在发生相变时的一个常见的现象,它通常涉及到新相的形成及两相界面的移动。这对于进行大离子半径的Na+脱嵌的层状氧化物更为明显,从而导致其出现严重的局部应变、裂纹和容量衰减。此外,相变过程产生的晶格畸变严重影响了氧化物正极的电子结构和氧化还原反应,进一步影响电极材料的电化学行为。据报道,在LiFePO4相变过程中检测到的亚稳态的固溶体能显著提高其倍率性能,但这种亚稳态的固溶体在低电流倍率下会弛豫为两个具有不同Li含量的相。这种非平衡的亚稳态中间相在钠电层状氧化物中也有相关报道,能够显著提高钠电层状氧化物的倍率性能和循环稳定性。但是,它们弛豫或者小电流倍率充放电情况下,会自发地转化为两个热力学有利的富Na相和贫Na相,随之而来的相界面会阻碍Na+的传输。这种相变过程中的非平衡固溶路径目前仅在高电流倍率充放电过程中被证实,在低电流倍率下的平衡固溶途径很少被报道。因此,通过调节钠电层状氧化物的相变途径,有望抑制相界面的产生,加速Na+的扩散,进而提高其倍率性能和循环稳定性。
二、研究工作简单介绍(请表述本工作的核心观点)
近日,南开大学李福军研究员团队报道了一种钠电层状氧化物正极P’2-Na0.653Ni0.081Mn0.799Ti0.120O2 (P’2-Na0.653NMT)。通过在TMO2层中掺入Ni和Ti,增强了O(2p)-Mn(3d-eg*)轨道杂化,抑制了MnO6八面体的Jahn-Teller扭曲,并引起了TMO2层收缩和Na层扩大。高电压下的P’2-Na0.348NMT到OP4-Na0.104NMT的稳态固溶体路径阻止了两相反应界面的产生,减小了相变过程的能垒,加速了Na+的扩散,实现了钠电层状氧化物正极优异的倍率性能和循环稳定性。该研究以“Homeostatic Solid Solution in Layered Transition-Metal Oxide Cathodes of Sodium-Ion Batteries”为题发表在Journal of the American Chemical Society上。文章第一作者为南开大学博士生任猛。
二、 核心内容表述部分
作者通过将Ni和Ti引入P'2-Na0.649MnO2 (P'2-Na0.649M)中,得到了层状氧化物正极P’2-Na0.653Ni0.081Mn0.799Ti0.120O2 (P’2-Na0.653NMT)。图1a-c证实了其为正交P’2结构,空间群为Cmcm,氧层的排列方式为ABBA,Ni、Mn、Ti同处于过渡金属层且呈无序状排列。二次离子质谱 (SIMS) 揭示了P’2-Na0.653NMT中各元素的均匀分布。随着Ni和Ti的引入,P’2-Na0.653NMT的DOS中费米能级的左移和O (2p)的右移说明了O (2p)-Mn (3d-eg*)杂化作用的增强(图1e和f)。图1g中P'2-Na0.653NMT的Mn K边的右移也反映了Mn和O之间成键作用的增强,对应于Mn-O键长变短(图1h)。结合COHP分析,P'2-Na0.653NMT中Mn-O键的共价性提高,进一步表明了Mn 3d和O 2p之间轨道杂化作用增强。
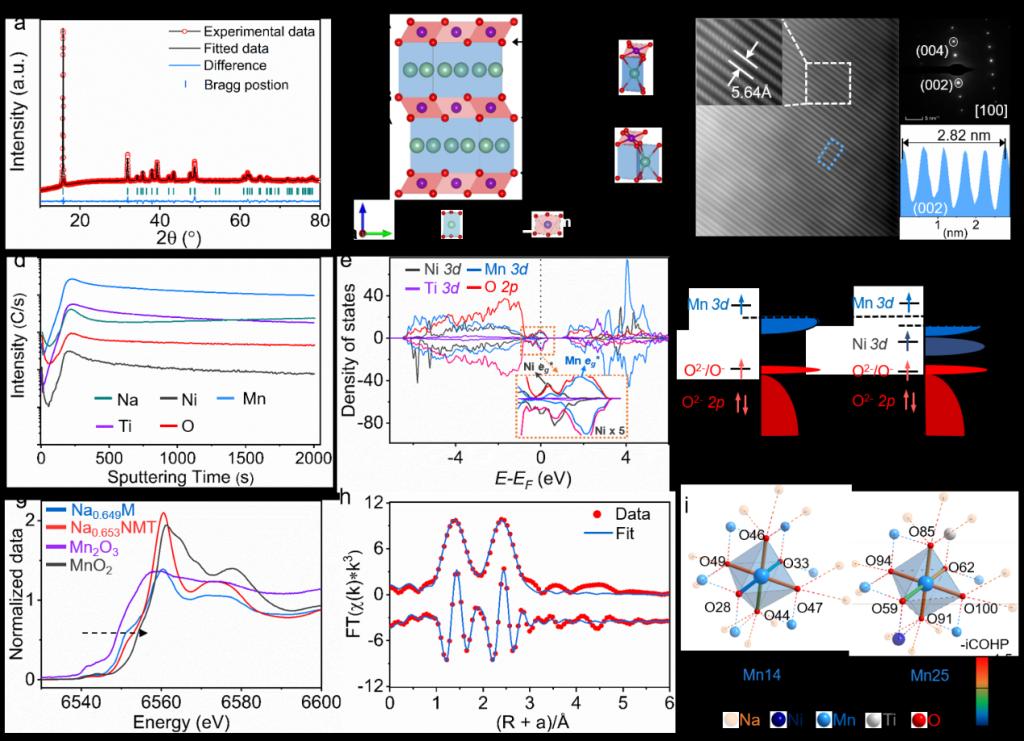
Figure 1. 结构分析。(a) P’2-Na0.653NMT的XRD图谱和精修;(b) P'2型结构;(c) HADDF-STEM图像,电子衍射图案和(002)晶面的晶格间距;(d) SIMS图谱;(e) P’2-Na0.653NMT的DOS;(f) P'2-Na0.653NMT和P'2-Na0.649M的电子结构和(g)XANES光谱;(h) P'2-Na0.653NMT的Mn K边EXAFS和在R空间的拟合;(i) P'2-Na0.653NMT和P'2-Na0.649M的COHP分析。
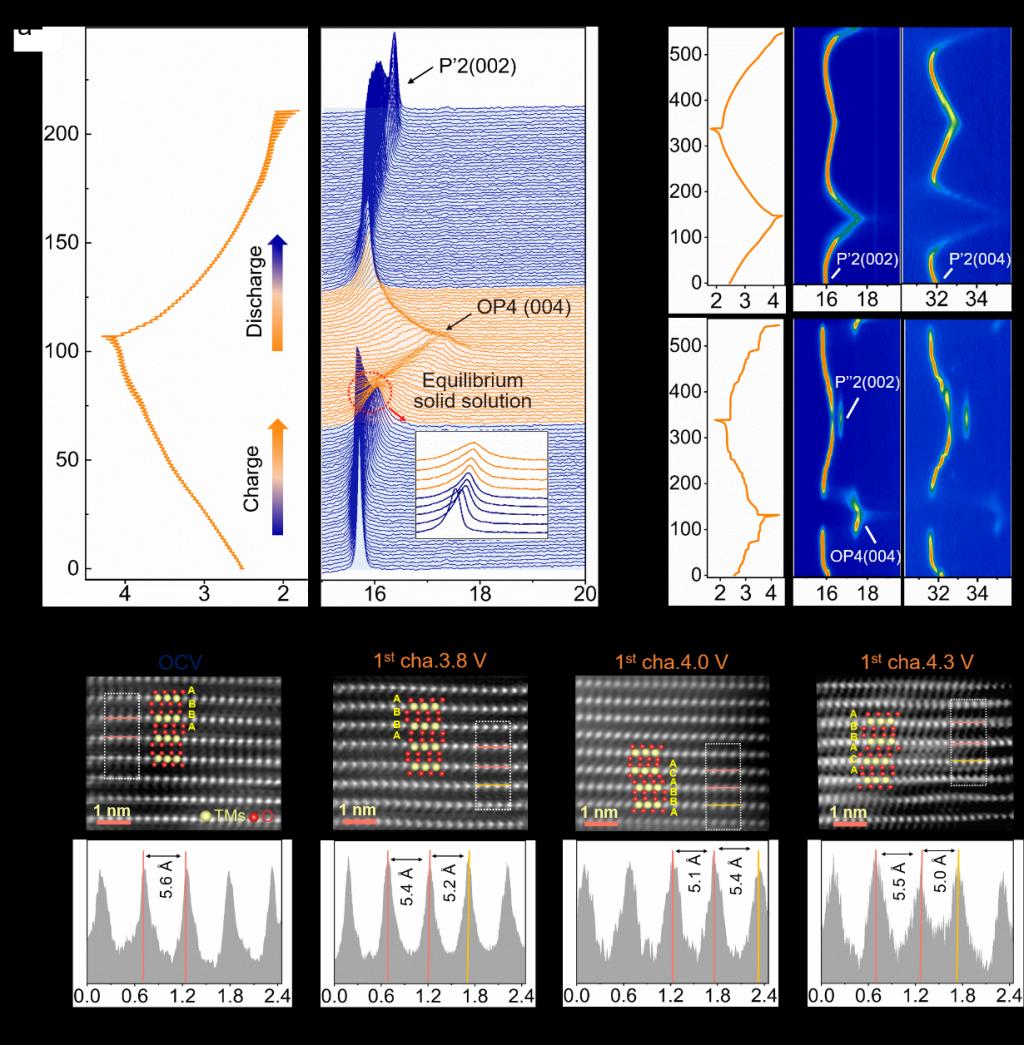
Figure 2. P'2-Na0.653NMT和P'2-Na0.649M的结构演变。(a) 以5 mA g-1间歇性充电/放电0.5小时和弛豫1小时的GITT曲线,以及P'2-Na0.653NMT的原位XRD图谱;(b) P'2-Na0.653NMT和(c) P'2-Na0.649M的原位XRD图谱及其等高线图;在(d)OCV、(e)3.8V充电、(f)4.0V充电和(g)4.3V充电状态下的HAADF-STEM图像。
图2中通过原位XRD和球差电镜等分析手段探究了P'2-Na0.653NMT在充放电过程中的结构变化。利用GITT和原位XRD证明了P'2-Na0.653NMT中出现了一种连续固溶反应路径,且在弛豫过程中,这种固溶反应不受自发的动力学反应的影响,表明其是热力学稳定的。同样的固溶反应在图2b的恒流充放电条件下测试的原位XRD图谱中被证实。图2d-g揭示了P'2-Na0.653NMT在不同充放电状态下的结构变化,在OCV和充电至3.8 V的情况下,其表现出P'2相的结构特征。充电至4.0及4.3 V,由于过渡金属的滑移,出现了P'2到OP4的相转变,但没有明显的相界面出现。上述结果证实了P'2-Na0.653NMT的相变是通过一种稳态的固溶反应路径进行的。

Figure 3. 模拟的相变过程。(a) P’2-Na0.653NMT的相图和(b) 相应的G.S.结构示意图;(c) P′2- Na0.649M和(d) 相应的G.S.结构示意图;(e) 从P′2到OP4相的三种反应途径的示意图:稳态固溶体、非平衡固溶体和两相反应。
作者基于第一性原理计算对P’2-NaxNMT的相变过程进行模拟。当0.062 < x < 0.25时,P’2-NaxNMT的不同基态 (G.S.) 结构能量之间的差异较小,使得P’2-NaxNMT和OP4-NaxNMT 相之间发生固溶成为可能 (图3a和b)。在0.187 < x < 0.5时,P’2-NaxM是由P’2-Na0.5M和OP4-Na0.187M的G.S.混合结构组成,且两相在每个G.S.的形成能差异较大 (图3c和d)。这种大的能量差异导致其相结构在能量上和空间上的分离,进而诱发了典型的两相反应。
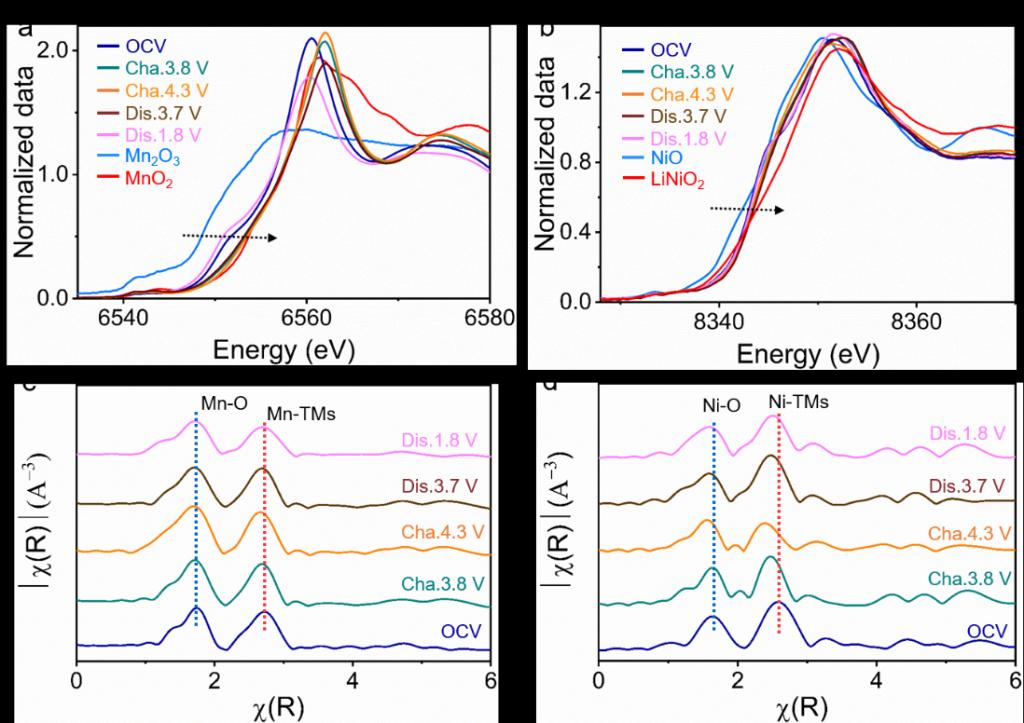
Figure 4. P’2-Na0.653NMT的电荷补偿机制。在不同的充电/放电状态下收集的(a) Mn K-边和(b) Ni K-边的XANES图谱;相应的(c) Mn K-边和(d) Ni K-边的傅里叶变换EXAFS光谱。
图4中揭示了P’2-Na0.653NMT在充放电过程中的电荷补偿机制。从OCV充电至3.8 V,Mn离子参与电荷补偿,Mn3.37+被氧化为Mn3.98+。继续充电至4.3 V,Ni离子参与电荷补偿,Ni2.08+被氧化为Ni2.66+。这与图1e中的DOS结果相一致,Mn要先于Ni发生氧化还原反应参与P’2-Na0.653NMT的电荷补偿过程。
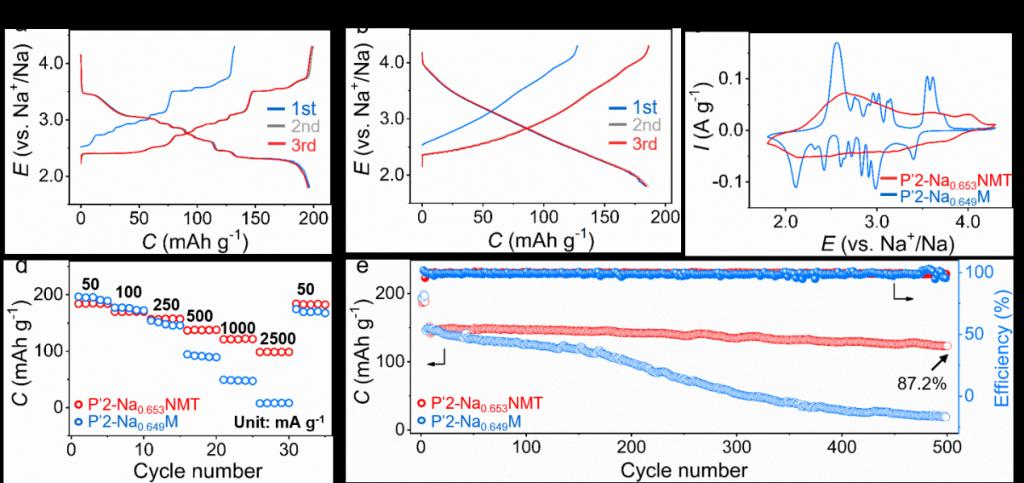
Figure 5. P’2-Na0.649M和P’2-Na0.653NMT的电化学性能。(a) P’2-Na0.649M和(b) P’2- Na0.653NMT在1.8至4.3V范围内,25 mA g-1的充电/放电曲线;(c) 第二周期在0.1 mV s-1的CV曲线;(d) 倍率性能;(e)在250 mA g-1下的循环性能。
图5中对P’2-Na0.649M和P’2-Na0.653NMT的电化学性能进行了评估。图5a和b展示了它们在第二个循环中的充/放电曲线。P’2-Na0.649M的放电容量达到198 mA h g–1,相当于嵌入0.75 mol Na+;P’2-Na0.653NMT的容量略有下降,达到185 mA h g–1,相当于嵌入0.70 mol Na+。P’2-Na0.649M的充/放电曲线出现多个平台,这源于Na+/空位重排以及分别在3.5和2.2 V发生的P’2 ↔OP4和P’2 ↔ P’’2两相反应。P’2-Na0.653NMT的充/放电曲线是平滑的,没有出现任何平台。图5c所示的循环伏安(CV)曲线进一步证实了P’2-Na0.653NMT在脱钠和嵌钠过程中的固溶反应途径。这显著提高了P’2-Na0.653NMT的倍率性能 (图5d) 和循环稳定性 (图5e,500圈后的容量保持率为87.2%)。
结论
综上所述,作者合成了一种层状氧化物正极P’2-Na0.653NMT。由于Ni和Ti掺入了TMO2 层, O(2p)-Mn(3d-eg*)的轨道杂化增强,使得TMO2 层收缩,Na层间距扩大,MnO6八面体的Jahn-Teller畸变得到缓解。结构稳定性增强带来的相变能垒的降低诱导了高电压下P’2-Na0.348NMT到OP4-Na0.104NMT的稳态固溶转变。同时,低电压下的P’2 ↔ P’’2的相变也受到抑制。这种全电压范围的单相转变途径加快了P’2-Na0.653NMT的Na+传输,使其表现出出色的倍率性能和优异的循环稳定性。目前,在层状氧化物中,相变过程的稳态固溶途径很少被报道,这将为获得高性能钠离子电池正极材料提供参考。
四、文献详情
Homeostatic Solid Solution in Layered Transition-Metal Oxide Cathodes of Sodium-Ion Batteries
Meng Ren, Shuo Zhao, Suning Gao, Tong Zhang, Machuan Hou, Wei Zhang, Kun Feng, Jun Zhong, Weibo Hua, Sylvio Indris, Kai Zhang, Jun Chen and Fujun Li*
J. Am. Chem. Soc. 2022, DOI: 10.1021/jacs.2c09725