研究背景
金属Na具有较低的还原电位(-2.71 V vs. SHE)和较高的理论比容量(1166 mAh g-1),受到研究人员的广泛关注。然而,在重复的沉积/剥离过程中,Na负极较高的反应活性导致其与传统电解液反应生成不稳定的固体电解质界面膜(SEI),加剧负极表面Na+离子流的不均匀分布,进而诱发不受控制的枝晶生长。而Na枝晶所导致的界面高比表面积不可避免会导致不稳定SEI膜的破裂与累积,消耗大量活性钠,降低钠的利用率。同时,SEI膜的积累会增大钠离子的传输能垒,导致缓慢的界面动力学。因此,精细调控界面反应,生成均匀稳定且坚固的SEI膜是非常有必要的。近年来,阴离子衍生的SEI因其具有机械强度高、离子电导率高、离子扩散快等特点,在提升电池的电化学性能等方面展现出极大的优势。
阴离子的溶剂化能力是形成阴离子衍生SEI膜的关键,它高度依赖于溶剂-阳离子和阴离子-阳离子之间的竞争配位。通过引入弱溶剂化的溶剂或强配位能力的阴离子,可实现常规浓度下更多阴离子参与溶剂化构型配位,一般来讲,这类电解液中盐的解离度低,离子电导率偏低。高浓度电解液(HCE)体系中,受缺乏可用溶剂的调控,阴离子参与Na+周围的第一溶剂化鞘层。然而,由于粘度高、浸润能力差以及成本高昂,HCE的应用受到一定限制。最近,一种新型局部高浓度电解液(LHCE)的概念被广泛应用,在HCE中加入惰性的氢氟醚共溶剂,可有效缓解上述问题。尽管其电化学性能有显著的改善,但惰性稀释剂的引入对电解液中局部Na+配位结构及配位环境的影响仍缺乏深入细致的探索。
成果简介
在此,南开大学李福军研究员团队,近日在Angew. Chem. Int. Ed.期刊上发表题为“Anion Reinforced Solvation for Gradient Inorganic-Rich Interphase Enables High-Rate and Stable Sodium Batteries”的研究性论文。文中以摩尔比NaFSI:DME:OTE(1H,1H,5H-八氟戊基-1,1,2,2-四氟乙基醚)=1:1.5:3的LHCE为模型电解液,在钠负极表面原位构建梯度分布的富无机SEI膜。在OTE存在下,电解液的溶剂化构型由HCE中的三维网络聚集转变为LHCE中的阴阳离子溶剂团簇,促进更多的阴离子进入第一溶剂化鞘层。阴阳离子溶剂团簇在电解液中的整体迁移机制导致大量阴离子富集在Na表面,促进FSI-阴离子的逐步分解,从而形成富含无机且梯度分布的超薄SEI,如NaF、NaSON、Na2S等,进一步降低了Na+在SEI膜中传输的活化能。这些优点使Na||Na3V2(PO4)3电池具有优异的倍率性能和长循环稳定性。这项工作为调控电极-电解液界面膜助力高能量密度钠金属电池发展提供了新的见解。
图文解析

图1.(a)各电解液及溶剂的Raman谱图;(b)自由DME和配位DME的占比;(c)径向分布函数和配位数;(d)最具代表性的Na+配位环境;(e)溶剂化结构快照。
要点:
1. 加入OTE后,FSI-的伸缩振动峰从738 cm-1上升至745 cm-1,配位DME的占比从78.2%降低至64.6%,意味着更多FSI-阴离子参与内溶剂化鞘层。
2. HCE和LHCE中内溶剂化鞘层中均被DME分子和FSI-阴离子占据,引入OTE后,Na+-FSI-之间的距离从2.48 Å降低至2.34 Å,同时,Na+-DME之间的距离从2.32 Å上升至2.35 Å,意味着增强的Na+-FSI-相互作用力。
3. Na+-OTE之间的距离为5.97 Å,表明稀释剂不参与Na+内溶剂化鞘层。
4. HCE中参与Na+溶剂化配位的FSI-与DME的比值为1.00:1,该比值在LHCE中上升为1.08:1,即LHCE中有更多的阴离子及更少的溶剂参与内溶剂化鞘层。
5. 引入OTE导致更多阴离子参与溶剂化配位的原因:OTE完全不溶NaFSI盐,可以和DME互溶。HCE中加入大量OTE,可导致生成更多被OTE包围不能溶解钠盐的自由DME,降低配位DME的比例,即增大配位FSI-的比例。
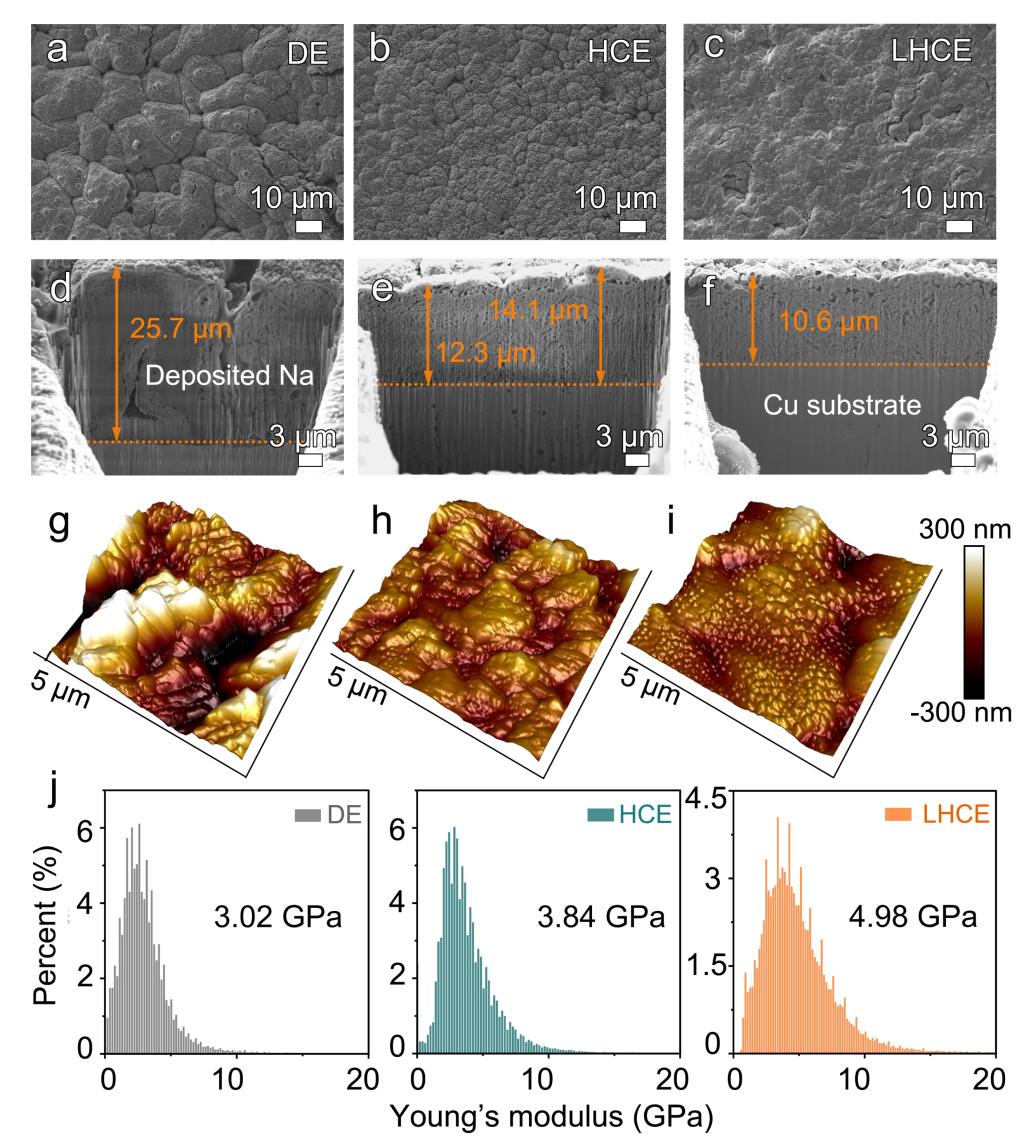
图2.各电解液中电沉积Na的(a-c)平面及(d-f)截面FIB-SEM图像;各电解液中电沉积Na的(g-h)三维AFM图像及(j)相应的杨氏模量柱状图。
要点:
1. 加入OTE后,电沉积Na的均匀性及致密度得到明显改善。
2. 加入OTE后,机械强度从3.84 GPa提升至4.98 GPa,增强的SEI机械模量在抑制枝晶生长、诱导钠均匀沉积方面起着关键作用。
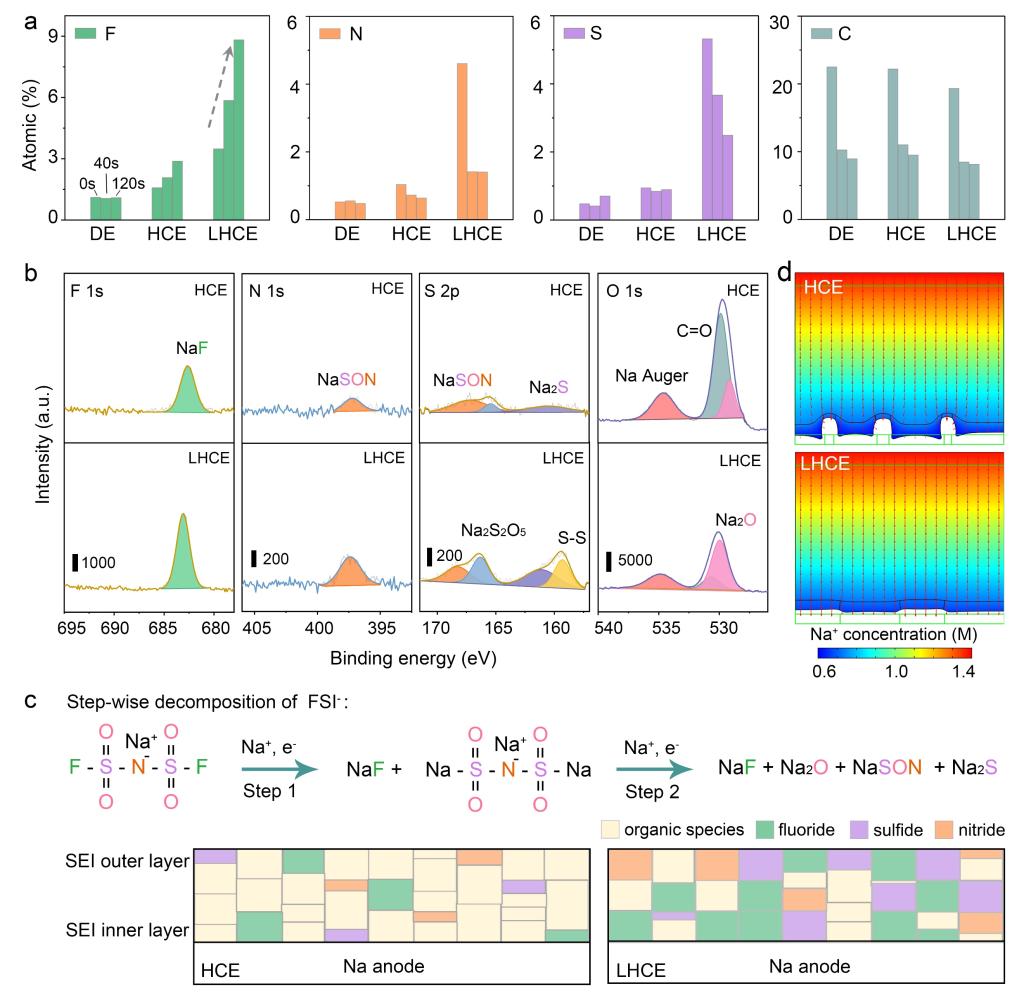
图3.(a)各电解液中Na负极表面SEI膜中F、N、S、C的原子比,刻蚀时间为0,40和120 s;(b)刻蚀40 s后各电解液中F 1s, N 1s, S 2p 和O 1s的XPS谱;(c)FSI-阴离子的分解路径及SEI组成示意图;(d)钠沉积行为的有限元模拟。
要点:
1. LHCE中呈现梯度分布的富无机SEI膜,其中氟化物主要分布在SEI内层,硫化物和氮化物主要分布在SEI外层,有机组分则与传统SEI的分布相似,主要分布在SEI膜的外层。这主要归因于钠负极表面大量FSI-阴离子的逐步分解,具体过程见FIG 3c分解示意图。
HCE体系的SEI中无机组分呈现相对均匀的分布,归因于钠负极表面FSI-阴离子数量少,同时发生分解路径中的步骤1和2。
2. 不同电解液体系中迁移至钠负极表面FSI-阴离子数量不同的原因:HCE中Na+以跳跃机制在三维网络状的聚集体中传输,根据Stokes-Einstein方程计算得到,LHCE中的流体力学半径为0.93 nm,远远大于Na+半径,意味着在LHCE中Na+以阴阳离子溶剂团簇的形式整体迁移,因此有大量的阴阳离子溶剂团簇迁移至Na负极表面并去溶剂化生成FSI-阴离子。
3. LHCE中FSI-分解更完全:HCE中较弱的分解产物(161.1 eV处的Na2S,529.5 eV处的Na2O,N 1s谱中397.5 eV和S 2p谱中168.3 eV处的NaSON),在LHCE中均展现更强的峰。同时,中间产物Na3S2O4N的分解比例从HCE中的73.4%提升至86.9%。
4. LHCE中SEI的无机组分含量是HCE中的2-3倍的原因:除却更多的阴离子参与溶剂化构型配位,更主要的原因为稀释剂的引入改变了溶剂化配位环境,改变Na+在电解液中的传输方式,促进阴离子的完全分解。

图4.(a)中间SEI膜被刮除后的AFM图像;(b)SEI膜的厚度;(c)交换电流密度;Na||Na3V2(PO4)3电池的(d)倍率性能和(e)长循环。
要点:
1. SEI膜均匀性的测试:以金镀层的平板电极为集流体搭建电化学池,在其上原位生成SEI膜,并通过AFM测试其均匀性。可发现HCE和LHCE体系中SEI膜均由均匀分布的颗粒状产物堆积而成,其平均粒径分别为120.2 nm和101.8 nm,相应的表面粗糙度分别为8.5 nm和6.1 nm。
2.SEI膜厚度的测试:ScanAsyst-air模式无损采集SEI膜表面形貌(5×5 μm2),Contact模式施加500 nN的力来回刮擦去除SEI膜(2.5×2.5 μm2),最后采用ScanAsyst-air模式采集形貌(5×5 μm2),对其进行截面高度分析可得到SEI膜的厚度。LHCE中检测到其具有超薄的SEI膜,厚度仅为HCE体系的1/3(4.33 nm vs. 13.37 nm)。
3. 得益于超薄富含无机组分SEI膜的建立,LHCE中具有更快的界面动力学速率。Na||Na3V2(PO4)3电池中展现出优异的倍率性能(24 C下79.9 mAh g-1的容量,1C=110 mA g-1)以及1C下循环6000圈后94.2%的高容量保持率。
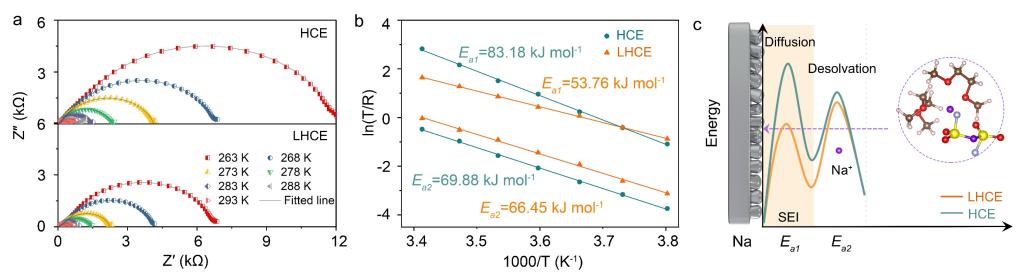
图5.(a)变温对称电池的奎因斯特图;(b)Na+穿过SEI膜的活化能)(Ea1)和Na+去溶剂化的能量(Ea2);(c)Na+沉积界面行为示意图。
要点:
1. 测试变温Na||Na对称电池的电化学阻抗谱并对其进行拟合。高频区的半圆对应Na+穿过SEI膜的阻抗,低频区的半圆对应电荷转移阻抗,即Na+去溶剂化过程。可根据Arrhenius方程计算得到相应的活化能。
2. 得益于LHCE中超薄致密且富含无机组分的SEI膜,其具有较多且快速的离子传输通道,Na+穿过SEI膜所需的能量(Ea1)大幅降低(53.76 kJ mol-1 vs. 83.18 kJ mol-1)。
3. 得益于LHCE中更多的阴离子参与溶剂化构型配位,其去溶剂化能也有一定程度的降低(64.09 kJ mol-1 vs. 69.88 kJ mol-1)。
文章链接
Anion reinforced solvation for gradient inorganic-rich interphase enables high-rate and stable sodium batteries, Xunzhu Zhou, Qiu Zhang, Zhuo Zhu, Yichao Cai, Haixia Li, Fujun Li*. Angewandte Chemie International Edition, 2022, DOI: 10.1002/anie.202205045.
网址:
https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.202205045