袁明鉴课题组Angew:电催化CO2还原为CH4的石墨炔调节的Cu单原子催化剂中Cu-C键的构建
来源:原位人

1、摘要简介
通过精心设计催化剂来调控中间体以控制反应方向是提高电催化CO2转化为CH4选择性的关键。M−C (M=金属)键对于调节多电子反应尤为重要。然而,它在纳米材料中的构建是具有挑战性的。本研究通过合理设计Cu SAs(单原子)在石墨炔上原位锚定,首次实现了Cu−C (GDY)化学键的构建。原位拉曼光谱电化学和DFT计算证实,由于Cu−C键的形成,在CO2还原过程中,Cu原子上主要形成的中间体是*OCHO,而不是*COOH,这有利于CH4的形成。因此,我们发现在Cu SAs/GDY中构建Cu−C键可以提供有效的电荷转移通道,但最重要的是控制反应中间体,引导更容易的反应途径将CO2转化为CH4,从而显著提高其催化性能。这项工作为在原子水平上提高CO2RR的选择性提供了新的见解。
2、背景介绍
电催化还原CO2制烃类燃料是一种极具吸引力的策略,通过与可再生能源发电相结合来实现碳中和能源循环。CO2还原反应(CO2RR)本质上是一个多质子耦合的电子转移过程。随着转移电子和质子数量的增加,反应路径变得越来越复杂模糊。由于开发单碳产物CH 4需要的电子转移数最多,8电子,因此探索CO2对CH4的电催化还原过程对于了解CO2RR的本质具有重要意义。实际上,非贵金属过渡金属铜(Cu)是迄今为止催化CO2转化为多电子还原产物最可靠的金属材料。要实现CH4在Cu催化剂上的演化,必须解决两个关键问题。一是调节反应中间体或抑制碳-碳偶联反应的发生,避免生成CO或其他多碳产物。二是加速由8个电子转移步骤引起的缓慢的动力学过程,从而促进CH4的生成。然而,这两个棘手的问题很难同时解决。此外, Cu催化剂的形貌、氧化态和晶格面对CO2RR的选择性有显著影响。因此,在铜基电催化剂体系中实现CH4的高选择性仍然是一个巨大的挑战。
构建铜单原子催化剂似乎是实现上述目标的理想途径。由于催化位点的单分散,可以有效抑制C−C偶联反应,从而避免多碳产物的形成。然而,单分散铜的单链反应往往不足以有效地切断其他单碳产物的反应路径,特别是CO。CO2分子的活化对产物的选择性起着重要作用,生成的*CO中间体及其随后的质子化过程导致形成各种单碳产物,从而降低了选择性。近年来的理论研究和非铜基实验结果表明,改变CO2分子的活化位点,即生成O原子优先成键的中间体(*OCHO),可以有效阻断CO反应路径,促进CH4的形成。然而,如何在Cu SAC体系中实现这一目标,是一个由Cu原子固有属性引起的难题。金属位点与载体之间的电子调节效应已被证明可以调节金属原子的电子结构,从而影响关键中间体的吸附行为。因此,需要一种理想的在载体材料,既能稳定单个铜原子,又能有效增强电子调控效应。
石墨炔(Graphdiyne, GDY)是一种由sp2-和sp-杂化碳原子组成的新型碳同素异形体。GDY中独特的-C≡C-C≡C-结构可以有效地稳定单原子,并大大促进GDY与原子中心之间的电荷转移。因此,它可以导致单原子电子环境的变化。此外,理论计算表明,由于GDY的多孔结构,GDY具有良好的CO2吸附能力,这是后续还原反应的前提条件。这些特点使GDY成为合成和调节SAC的合适平台,特别是CO2RR中使用的SAC。此外,在有机金属配位化学中Cu−C键对调整有机反应方向是有用的。因此,可以利用Cu−C键来调节CO2转化为CH4的多电子反应。
3、图文解析
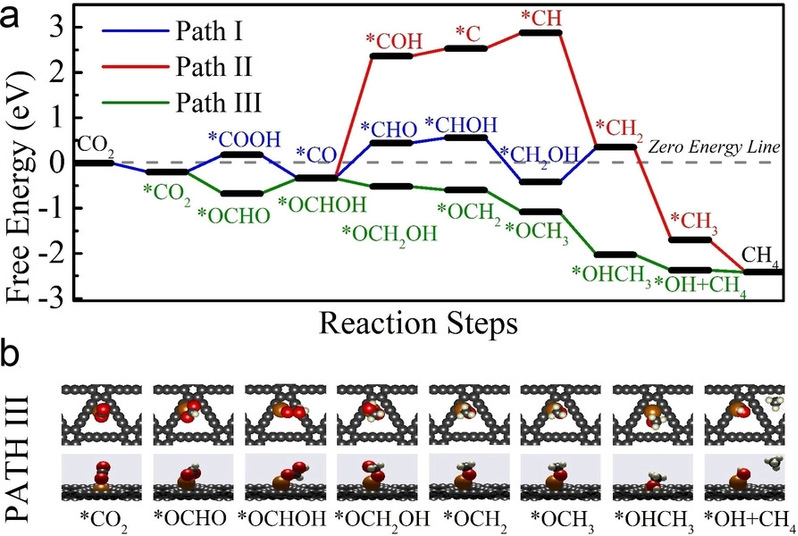
图1. Cu SAs/GDY上CO2RR→CH4的DFT计算。a)可能的反应路径和相应的零势能自由能图,H+和H2O省略。b) 路径 III中涉及的吸附物质的优化几何结构的俯视图和侧视图。O:红色,H:白色,C:灰色,Cu:橙色
为了充分揭示在Cu SAs/GDY上CO2RR到CH4的作用机理,进一步进行了与计算氢电极(CHE)模型结合的DFT计算。如图1a所示,考虑了三种可能的CH4生成途径。在第一次质子耦合电子转移反应中,路径III与路径I和路径II分离,即*CO2+(H++e−)→*COOH/*OCHO,其中*表示表面物种/位点。在图5b所示的优化几何构型中,可以明显看出*OCHO是CGDY-Cu−O*结合中间体,而*COOH是CGDY-Cu−C*结合中间体。*OCHO的自由能比*COOH低约0.88 eV,表明*OCHO在Cu SAs/GDY催化体系中比*COOH更稳定。为了了解*OCHO和*COOH之间不同的结合相互作用,研究了了*OCHO和*COOH的p-投影态密度(PDOS)对比图。O原子的p轨道和Cu原子的d轨道的重叠和共振表明在*OCHO中O和Cu原子之间存在强烈的轨道杂化。结果表明,在设计的Cu SAs/GDY上,*OCHO中间体比*COOH更容易形成。
测试结果分析
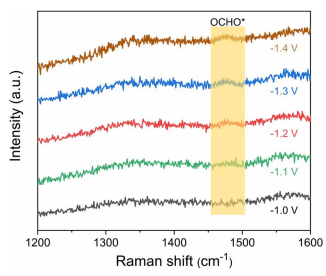
图2. Cu SAs/GDY在不同电位下的原位拉曼光谱
为了进一步验证理论计算得出的结论,获得了不同电位下的原位拉曼光谱(图2)。1475 cm−1附近的峰与OCHO*中间体有关。可以看出,在生成CH4的最佳电位下,OCHO*中间体的生成显著促进。这意味着Cu SAs/GDY上CO2向CH4的转化确实是通过OCHO*中间体进行的,这与DFT的结果是一致的。因此,PDOS结果和原位拉曼光谱强烈表明,设计的Cu SAs/GDY催化剂可以稳定*OCHO中间体。路径I和路径II接下来的质子和电子转移反应生成中间体*CO,它被认为是铜表面各种有机产物的关键中间体。然而,第二次质子-电子转移反应将路径III引向*OCHOH中间体。同时,*CO+H2O和*OCHOH的自由能大致相同。*CO生成CH4的反应有两条途径。在路径II中,氢与O结合形成*COH,自由能增加相对较高,为2.69 eV,表明路径II导致反应没有进展。路径1中,在*CO+(H++e−)→*CHO和*CH2OH+(H++e−)→*CH2+H2O中分别出现了0.77 eV和0.78 eV的较大自由能增加。这两个自由能垒将显著减缓反应动力学。相比之下,在路径III中,*OCHOH中间体之后的步骤是放热的,说明通过路径III生成CH4是平稳高效的。路径III的反应决速步RDS为*OCHO+(H++e−)→*OCHOH,自由能势垒为0.34 eV。由此总结CO2到CH4的反应路径如下:
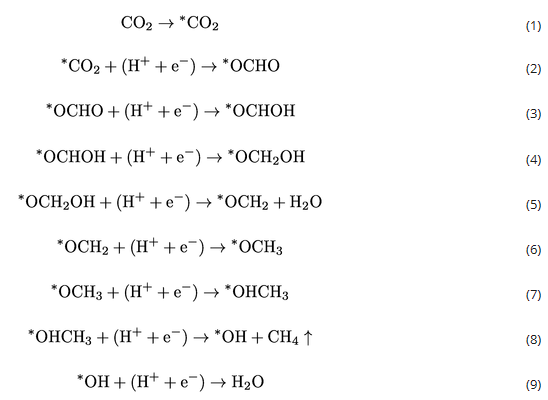
原位实验细节
原位电化学拉曼测试:用LabRAMHR拉曼光谱仪记录了原位拉曼光谱。采用H型流动原位池,在CO2饱和的0.1 M KHCO3水溶液中进行常压下原位拉曼测试。
4、结 论
该工作合理设计了一种高效稳定的CO2RR电催化剂,该催化剂具有丰富的锚定在GDY上的原子分散Cu单原子。首次发现GDY可以作为一种有效的载体材料,在Cu SAs和GDY之间建立Cu−C配位键,从而调节Cu单原子的电子环境。因此,它可以稳定Cu-Sas,防止其团聚,并为CO2到CH4的8电子还原过程提供有效的电荷转移途径。最重要的是,它还可以控制反应中间体,防止CO或其他多碳产物的生成,从而使CH4的反应路径更容易。Cu−C键被证实是CO2RR过程中形成*OCHO中间体的唯一原因。理论上证明*OCHO中间体有利于该体系中CH4的生成。结果表明,Cu SAs/GDY催化剂具有高达81%的CH4 FE和良好的稳定性。该工作将为非贵金属/GDY杂化材料在其他高效催化方面的应用提供新的见解。
原文链接:
Shi, G.; Xie, Y.; Du, L.; Fu, X.; Chen, X.; Xie, W.; Lu, T. B.; Yuan, M.; Wang, M., Constructing Cu-C Bonds in a Graphdiyne-Regulated Cu Single-Atom Electrocatalyst for CO(2) Reduction to CH(4). Angew Chem Int Ed Engl 2022, 61 (23), e202203569.
DOI: 10.1002/anie.202203569
https://doi.org/10.1002/anie.202203569